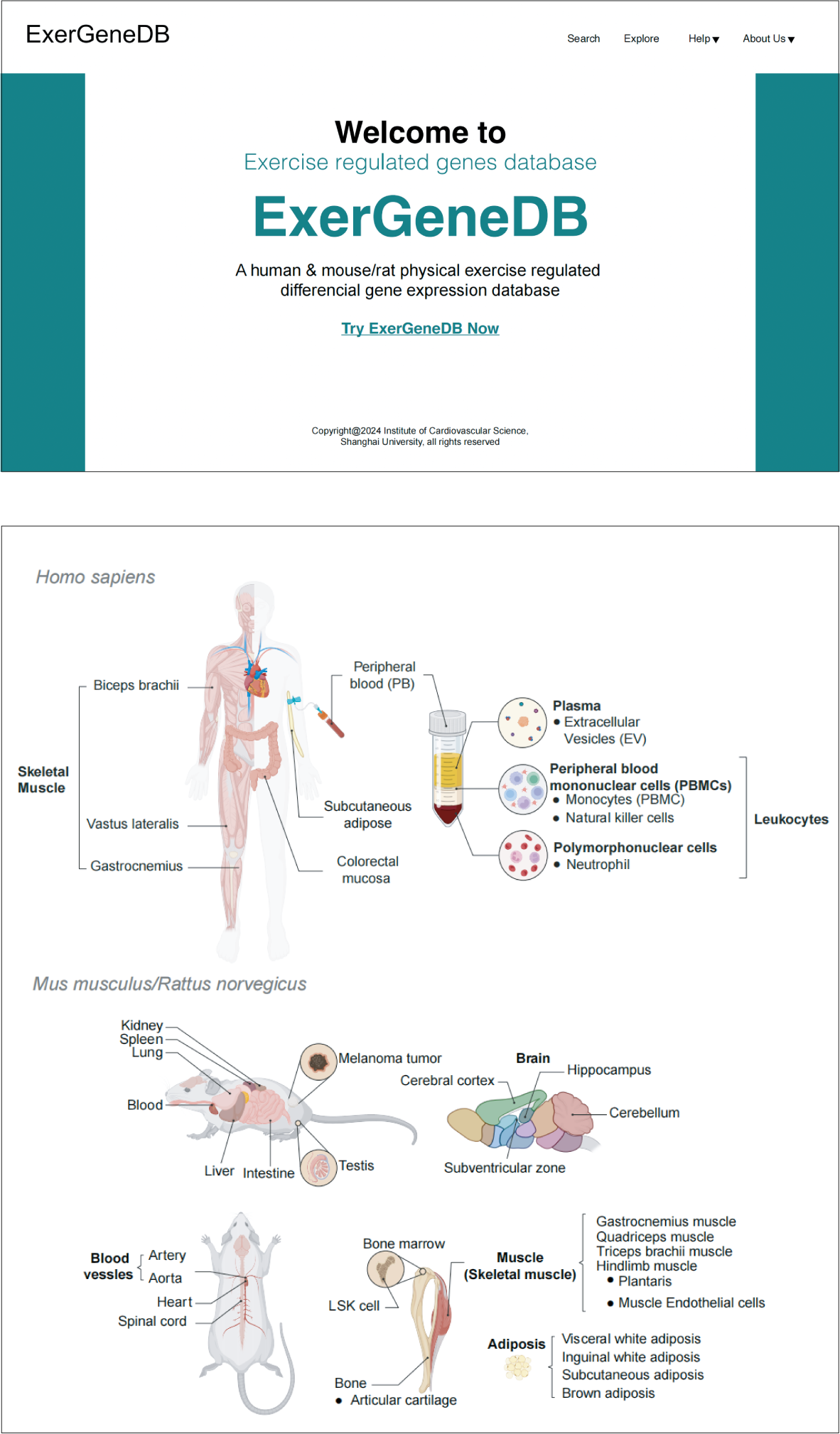
1 Data collecting
- Collect data from free public databases including NCBI and NGDC
- (Exercise[All Fields]) AND ("Homo sapiens"[Organism] OR "Mus musculus"[Organism] OR "Rattus norvegicus"[Organism]) Bibliographic retrival
2 Collate sample information
Sequencing information
Clinical information
Exercise information
3 Data process
High throughout RNA sequencing
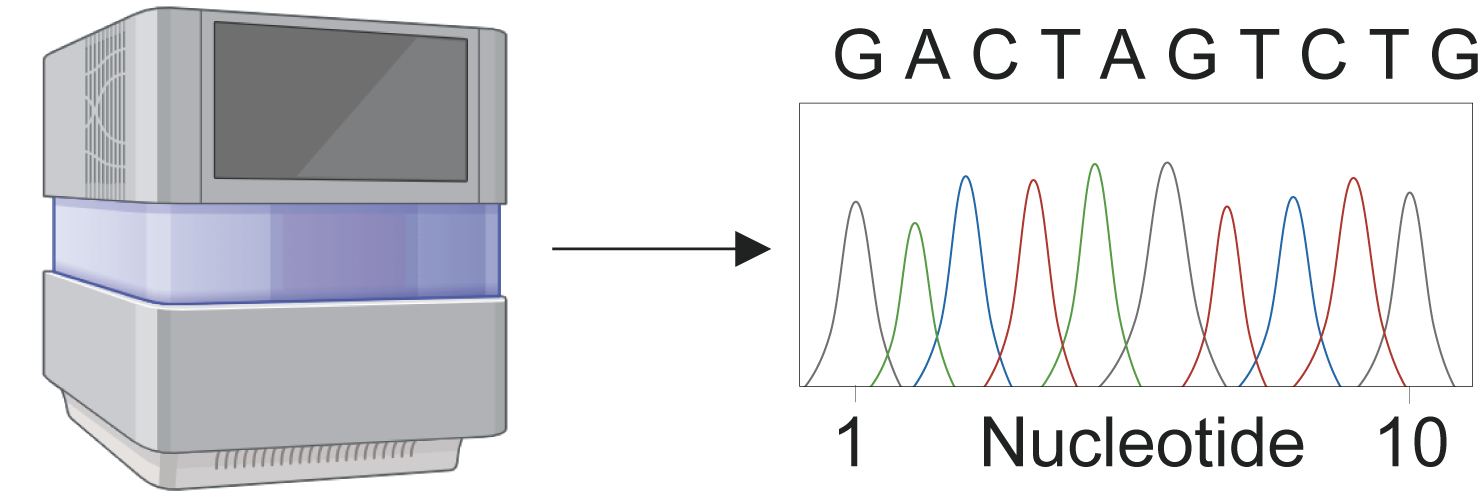
2 QC
FastQC(v0.12.1) is used to check sequence quality.
Trim_galore(v0.6.10) is used to apply adapter and quality trimming to fastq file.
1 Download data
Prefetch(v3.0.8) is used to download from free public database.
3 Quantification
HISAT2(v2.2.1) is used to align sequencing reads to reference genome.
FeatureCounts(v2.0.3) is used to transform reads to counts.
Expression profile by array
1 Download data
R package GEOquery (v2.66.0) is used to download raw data.
2 QC and Data process
Boxplot is used to check gene expression level.R package limma(v3.54.2)
backgroundCorrect() is used to filter background noise. R package DMwR2(v0.0.2) is used to impute NA values. Low and no expression genes are removed.
4 re-analysis DEG and build ExerGeneDB